![]()



![]()
snputils is a Python package designed to ease the processing and analysis of genomic datasets, while handling all the complexities of different genome formats and operations very efficiently. The library provides robust tools for handling sequencing and ancestry data, with a focus on performance, ease of use, and advanced visualization capabilities.
Developed in collaboration between Stanford University's Department of Biomedical Data Science, UC Santa Cruz Genomics Institute, and more collaborators worldwide.
snputils is stable and ready for production workflows. The core API is documented, tested, and suitable for day-to-day genomic analysis. The project is actively maintained: we ship regular releases, welcome contributions, and continue to extend format support, analyses, and performance.
- One API across genotype, local ancestry, global ancestry, phenotype, and IBD data
- Fast readers and writers for common population-genetics formats
- In-memory Python workflows and file-backed CLI workflows in the same package
- Ancestry-aware analyses including PCA and advanced alternatives, admixture mapping, and ancestry-specific allele frequencies
- Built-in plotting for embeddings, local ancestry, admixture, and association results
import snputils as su
snp = su.read_snp("cohort.vcf.gz") # VCF, BGEN, BED, PGEN
snp = snp.filter_biallelic_variants()
snp.save("cohort.pgen") # convert to PGEN
lai = su.read_lai("local_ancestry.msp") # MSP or FLARE local ancestry
adm = su.read_admixture("admixture_prefix") # ADMIXTURE-style global ancestry
pheno = su.read_pheno("phenotypes.tsv", col="trait")
ibd = su.read_ibd("segments.hapibd")
pcs = su.PCA(n_components=2).fit_transform(snp)
afr_af = snp.allele_freq(ancestry="AFR", laiobj=lai)
gwas = su.run_gwas(pheno, snp)
admix = su.run_admixture_mapping(pheno, lai)
su.viz.scatter(pcs, "labels.tsv", save_path="pca.png", show=False)
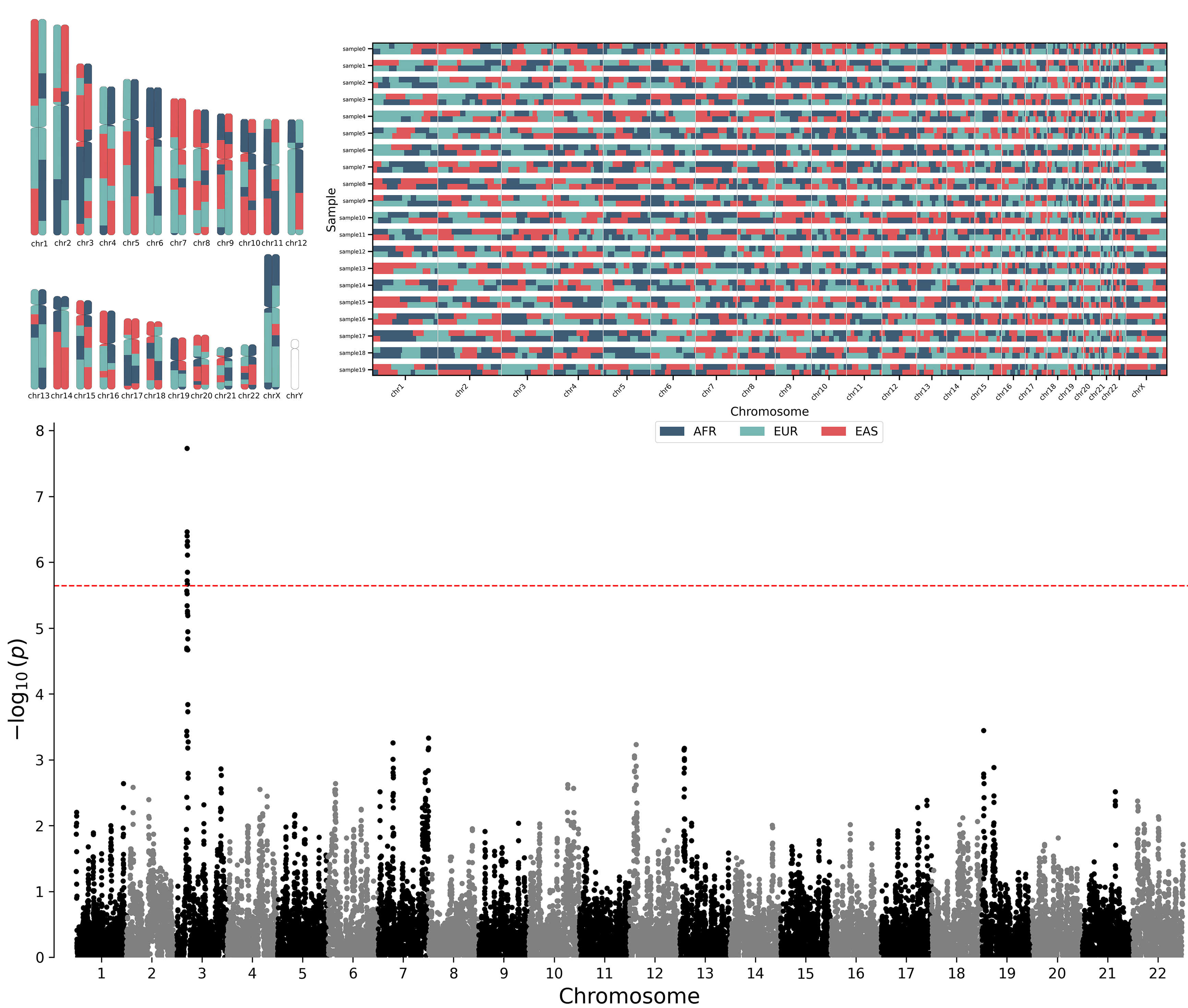
su.viz.chromosome_painting(lai, "chr_paintings/")
su.viz.qq_plot(gwas)
su.viz.manhattan_plot(admix)Basic installation using pip:
pip install snputilsOptionally, for PyTorch-backed features, install with the [torch] extra:
pip install 'snputils[torch]'Optional extras:
pip install 'snputils[tests]'for the test stackpip install 'snputils[docs]'for local documentation buildspip install 'snputils[demos]'for notebook demos
snputils provides high-level dispatchers like read_snp, read_lai, read_admixture, read_pheno, and read_ibd, plus explicit reader and writer classes when you need finer control.
- VCF: Support for
.vcfand.vcf.gzfiles - BGEN: Support for
.bgenfiles - PLINK1: Support for
.bed,.bim,.famfilesets - PLINK2: Support for
.pgen,.pvar,.psamfilesets - GRG: Read and write graph-based genome representation files
- Local Ancestry: Handle
.mspand FLARE.anc.vcf.gzlocal ancestry formats - Global Ancestry / ADMIXTURE: Read and write
.Qand.Pfiles - IBD: Read
hap-IBDandancIBDoutputs into a unified object
- SNPObject for genotype data, including filtering, saving, and allele-frequency helpers
- LocalAncestryObject and GlobalAncestryObject for ancestry-aware workflows
- PhenotypeObject, MultiPhenotypeObject, and CovariateObject for trait data
- IBDObject for segment filtering and ancestry-restricted trimming
- Synthetic dataset builders for SNP, mdPCA, maasMDS, chromosome-painting, admixture, and GRG examples
- Conversion helpers such as VCF-to-GRG workflows
-
Basic manipulation
- Filter variants and samples, correct SNP flips, and filter ambiguous SNPs
- Compute cohort and ancestry-specific allele frequencies via
SNPObject.allele_freq(...) - Stream allele frequencies with
snputils.stats.allele_freq_stream(...)for memory efficiency
-
Dimensionality reduction
- Standard PCA with optional PyTorch acceleration
- Missing-data PCA (
mdPCA) - Multi-array ancestry-specific MDS (
maasMDS)
-
Population-genetic statistics
- Compute
$D$ ,$f_2$ ,$f_3$ ,$f_4$ , the$f_4$ -ratio, and$F_{ST}$ (Hudson, Weir-Cockerham, and Tsallis$F_{q}$ ) - Block jackknife standard errors where applicable
- Optional ancestry masking in relevant workflows
- Compute
-
Association analysis
- GWAS on SNP dosages for binary and quantitative traits
- Admixture mapping from local ancestry dosage
- Built-in Manhattan and Q–Q plotting utilities
-
IBD and ancestry-aware trimming
- Unified IBD ingestion from common upstream tools
- Segment filtering and ancestry-restricted trimming using local ancestry
-
Simulation
- Lightweight haplotype-based simulation of admixed mosaics from founder haplotypes
- Scatter plots for PCA, mdPCA, and maasMDS embeddings
- Global ancestry bar plots
- Local ancestry visualization
- Chromosome painting
- Dataset-level cohort summaries
- Association plots
- Manhattan plots
- Q–Q plots
- Fast file I/O through built-in methods or optimized wrappers (e.g., Pgenlib for PLINK files)
- Memory-efficient operations using NumPy and Polars, including streaming workflows
- Optional GPU acceleration via PyTorch for computationally intensive tasks
- Support for large-scale genomic datasets through efficient memory management
Our benchmark demonstrates superior performance compared to existing tools:
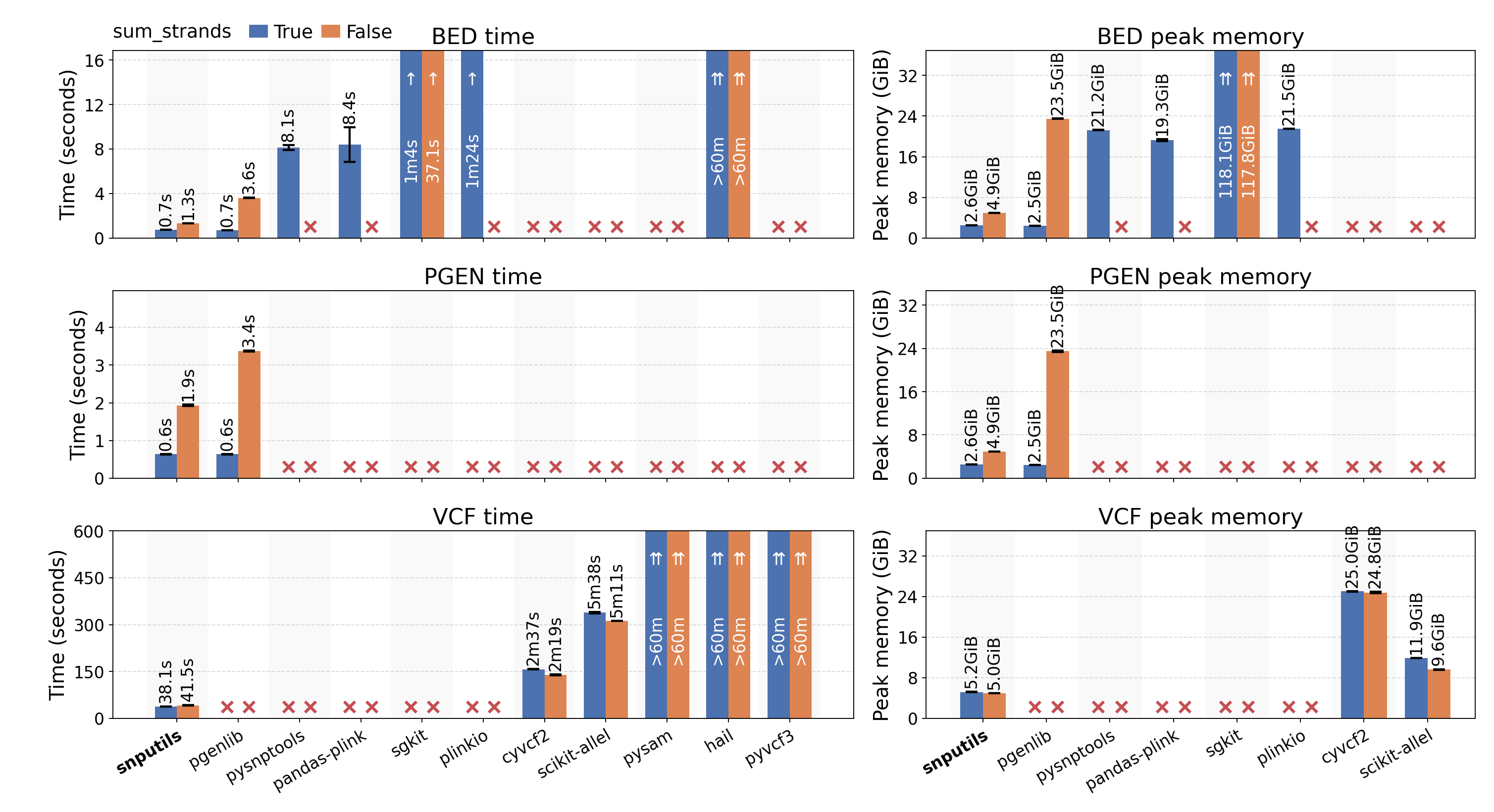
Reading time and peak-memory comparison for chromosome 22 data across different tools. See the benchmark directory for detailed methodology and results.
Installing the package provides a snputils command for common file-backed workflows:
snputils --help
snputils --versionAvailable subcommands include:
pca: run standard PCA and save coordinates/components and a scatter plot.mdpca: run missing-data PCA and save an embedding table.maasmds: run ancestry-specific MDS and save an embedding table.admixture-map: run admixture mapping from phenotype and local ancestry files.gwas: run variant-level association testing from phenotype and genotype files.simulate: simulate admixed haplotype batches from phased founder haplotypes.plot-manhattanandplot-qq: render association result visualizations.
The Python API remains the full surface for low-level readers/writers, object manipulation, IBD filtering and trimming, f-statistics, allele-frequency helpers, custom visualizations, and notebook-oriented workflows. Use the CLI when a workflow naturally starts from files and produces files; use Python when you need programmatic composition or in-memory objects.
- Documentation: docs.snputils.org
- Quickstart: Quickstart guide
- Tutorials: PCA, mdPCA, maasMDS, SNP objects, allele frequency, local ancestry visualization, admixture mapping, and GRG workflows
- API Reference: Readers, writers, data objects, processing classes, statistics, datasets, and visualization helpers
- Issues and feature requests: GitHub Issues
Top-level imports include:
- Readers and objects:
read_snp,read_lai,read_admixture,read_ibd,read_pheno,SNPObject,LocalAncestryObject,GlobalAncestryObject,IBDObject - Analysis:
PCA,mdPCA,maasMDS,run_gwas,run_admixture_mapping,allele_freq_stream - Datasets:
load_dataset,available_datasets_list,build_synthetic_* - Visualization namespace:
snputils.viz
If you use snputils in your research, please cite our paper:
@article{snputils2026,
author = {Bonet, David and Comajoan Cara, Marçal and Barrabés, Míriam and Smeriglio, Riccardo and Agrawal, Devang and Aounallah, Khaled and Geleta, Margarita and Dominguez Mantes, Albert and Thomassin, Christophe and Shanks, Cole and Huang, Edward C. and Franquesa Monés, Marc and Luis, Aina and Saurina, Joan and Perera, Maria and López, Cayetana and Sabat, Benet Oriol and Abante, Jordi and Moreno-Grau, Sonia and Mas Montserrat, Daniel and Ioannidis, Alexander G.},
title = {{snputils}: A High-Performance {Python} Library for Genetic Variation and Population Structure},
year = {2026},
doi = {10.64898/2026.02.28.708618},
url = {https://www.biorxiv.org/content/10.64898/2026.02.28.708618},
journal = {bioRxiv},
publisher = {Cold Spring Harbor Laboratory},
}We would like to thank the open-source packages that make snputils possible.